
Maladies
Rétinoblastome
Autres termes
- Gliome de la rétine
- Gliome rétinien
- Neuroblastome rétinien
- Rétinoblastome héréditaire
- Rétinoblastome familial
Définition
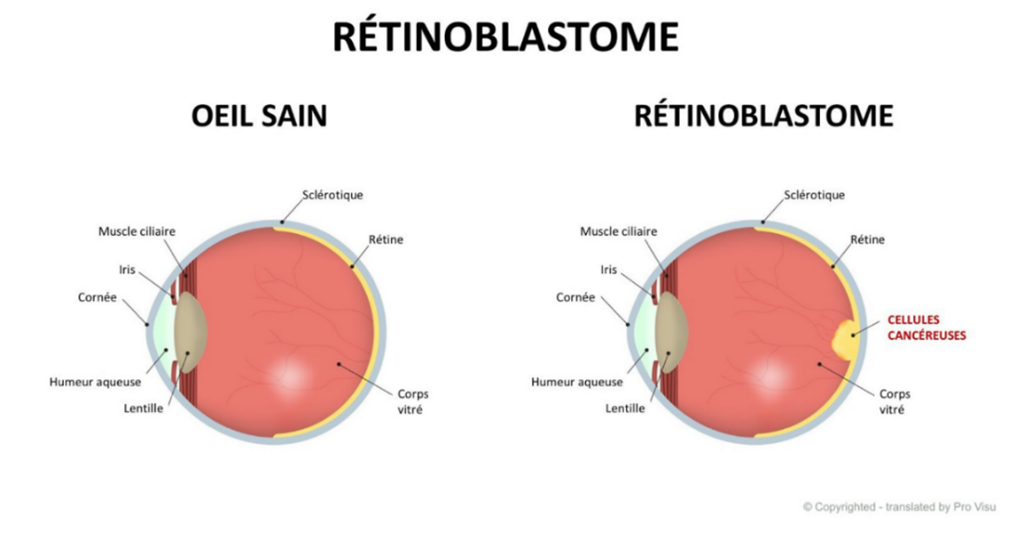
Le rétinoblastome est une tumeur maligne de la rétine qui se manifeste généralement jusqu’à l’âge de 5 ans, bien que la grande majorité des cas de rétinoblastome soit diagnostiquée dans les deux premières années de vie.
Il existe 3 types de rétinoblastomes :
- Non héréditaire, 60% des cas, touche un seul œil
- Héréditaire sporadique, 30% des cas
- Héréditaire familial, 10% des cas
Dans 70% des cas, le rétinoblastome est unilatéral (un seul œil atteint), bien que les enfants chez qui la maladie est héréditaire aient souvent une atteinte bilatérale. L’anomalie génétique qu’ils portent les prédisposent d’ailleurs au développement d’un autre cancer, c’est pourquoi ils doivent être régulièrement suivis.
Causes
Le rétinoblastome survient lorsque le gène du rétinoblastome, appelé RB1, est inactivé par une mutation ou une anomalie du chromosome 13 sur lequel il se situe. Dès lors, la division cellulaire devient incontrôlée, ce qui engendre la formation de tumeurs et détruit progressivement la vision.
Dans le cas d’un rétinoblastome non héréditaire, la mutation se fait lors de la réplication cellulaire qui a lieu après la conception. Pour le rétinoblastome héréditaire, c’est différent puisque la mutation est déjà dans les gamètes.
Symptômes
Le principal signe du rétinoblastome est la leucocorie, un reflet blanchâtre anormal de la pupille. Elle se remarque souvent de manière fortuite, par exemple sur des photographies qui ont été prises avec un flash, car le centre de l’œil est blanc plutôt que rouge.
Le strabisme peut également être un symptôme, bien que comme la leucocorie, il nécessite un examen de l’œil dans les sept jours pour confirmer le diagnostic et s’assurer qu’il ne soit pas lié à une autre pathologie oculaire.
Dans de rares cas, le rétinoblastome peut également être suspecté en cas d’inflammation oculaire, de baisse de la vision, de rougeur de l’œil ou de déformation de la pupille.
Diagnostic
Il est important que les parents et le pédiatre soient attentifs aux yeux des enfants dès leur plus jeune âge, car détecté précocement, le rétinoblastome a de bonnes chances de guérison. Plus le diagnostic est rapide, plus les risques de métastases et de dissémination dans les structures avoisinantes comme l’orbite, le nerf optique ou encore le cerveau (ce qui peut détruire la vision et engager le pronostic vital) sont réduits.
En cas de suspicion, il convient de réaliser un examen du fond de l’œil par ophtalmoscopie indirecte (les pupilles sont dilatées sous anesthésie générale) pour détecter des signes sur la rétine ou dans le vitré, puis de confirmer le diagnostic par une échographie, une IRM ou une tomodensitométrie orbitaire (TDM). Dans le cas d’une atteinte choroïdienne ou du nerf optique, une ponction lombaire ou une IRM cérébrale sont nécessaires pour rechercher d’éventuelles métastases.
Traitements
Plusieurs traitements sont possibles et ont pour objectif premier de préserver la vie de l’enfant. Le choix dépend de plusieurs facteurs :
- L’âge
- Le type d’atteinte (unilatérale ou bilatérale)
- La propagation ou non aux structures avoisinantes
- La présence ou non de métastases
- Les éventuels antécédents en matière de traitements
La chimiothérapie intra-artérielle de l’artère ophtalmique (chimiothérapie ciblée dans les vaisseaux sanguins qui irriguent la tumeur) et les traitements par laser ou cryocoagulation (gel de la lésion) peuvent suffire à détruire les petites tumeurs et à préserver la vision. Toutefois, lorsque la maladie est à un stade avancé, l’énucléation avec ablation d’une partie du nerf optique est parfois la seule option possible pour éviter la propagation des cellules cancéreuses.
Grâce aux traitements actuels, le taux de survie avoisine les 98%. De plus, lorsque le rétinoblastome est bilatéral, au moins un œil parvient dans la plupart des cas à être sauvé avec une vision fonctionnelle, même dans les cas avancés.
Fréquence
1 enfant sur 17’000 est touché par le rétinoblastome, ce qui représente entre 4 et 5 cas par année en Suisse. En général, ces enfants ont moins de 5 ans.
Le rétinoblastome est une pathologie rare, même s’il s’agit de la tumeur cancéreuse intraoculaire la plus fréquente chez les enfants puisqu’elle représente environ 4% des cancers pédiatriques. Les filles et les garçons sont autant susceptibles d’être touchés par le rétinoblastome.
Prévention
Il n’existe pas de moyen de prévention particulier, mais il convient de surveiller attentivement un enfant dont la famille a des antécédents de rétinoblastome. Les recommandations sont les suivantes : examiner l’enfant peu après sa naissance, puis 3 fois par an jusqu’à ses 4 ans.
Références
Rétinoblastome (RB) - Laboratoire d'oncogénomique - CHUV
Rétinoblastome - Hôpital ophtalmique Jules-Gonin
Rétinoblastome - Pédiatrie - Édition professionnelle du Manuel MSD (msdmanuals.com)
Bien vu !, le magazine de votre santé visuelle (N°8). Rétinoblastome : l’expertise lausannoise. Page 24, septembre 2022